Final (Revised) Common Rule — Part II
Exemption 5:
Public Benefit Service — Expanded with Changes (in bold below)
Research and demonstration projects that are conducted or supported by [Pre-2018 Rule Exemption 5 has been expanded to include federally-supported research. It is no longer limited to federally-conducted research] a Federal department or agency … and that are designed to study, evaluate, improve, or otherwise examine public benefit or service programs, including procedures for obtaining benefits or services under those programs, possible changes in or alternatives to those programs or procedures, or possible changes in methods or levels of payment for benefits or services under those programs. Such projects include, but are not limited to, internal studies by Federal employees, and studies under contracts or consulting arrangements, cooperative agreements, or grants. Exempt projects also include waivers of otherwise mandatory requirements using authorities such as sections 1115 and 1115A of the Social Security Act, as amended.
Each Federal department or agency conducting or supporting the research and demonstration projects must establish, on a publicly accessible Federal Web site or in such other manner as the department or agency head may determine, a list of the research and demonstration projects that the Federal department or agency conducts or supports under this provision. The research or demonstration project must be published on this list prior to commencing the research involving human subjects.
§ __.104(d)(5)
Exemption 6:
Taste and Food Evaluations — No Changes
§ __.104(d)(6)
Exemption 7:
Storage and Maintenance for Secondary Research — New Exemption
Storage or maintenance for secondary research for which broad consent is required: Storage or maintenance of identifiable private information or identifiable biospecimens for potential secondary research use if an IRB conducts a limited IRB review and makes the determinations required by § __.111(a)(8). [this is the new broad consent]
§ __.104(d)(7)
Exemption 8:
Information or Biospecimens in Secondary Research — New Exemption
Secondary research for which broad consent is required: Research involving the use of identifiable private information or identifiable biospecimens for secondary research use, if the following criteria are met:
- Broad consent for the storage, maintenance, and secondary research use of the identifiable private information or identifiable biospecimens was obtained in accordance with § __.116(a)(1) through (4), (a)(6), and (d);
- Documentation of informed consent or waiver of documentation of consent was obtained in accordance with § __.117;
- An IRB conducts a limited IRB review and makes the determination required by § __.111(a)(7) and makes the determination that the research to be conducted is within the scope of the broad consent referenced in paragraph (d)(8)(i) of this section; AND
- The investigator does not include returning individual research results to subjects as part of the study plan. This provision does not prevent an investigator from abiding by any legal requirements to return individual research results.
§ __.104(d)(8)

Improvements in Informed Consent
The prospective subject or the legally authorized representative must be provided with the information that a reasonable person would want to have in order to make an informed decision about whether to participate, and an opportunity to discuss that information.
[This is not applicable to broad consent] Informed consent must begin with a concise and focused presentation of the key information that is most likely to assist a prospective subject or legally authorized representative in understanding the reasons why one might or might not want to participate in the research. This part of the informed consent must be organized and presented in a way that facilitates comprehension.
Informed consent as a whole must present information in sufficient detail relating to the research, and must be organized and presented in a way that does not merely provide lists of isolated facts, but rather facilitates the prospective subject’s or legally authorized representative’s understanding of the reasons why one might or might not want to participate.
§ __.116(a)(4) and (5)(i)-(ii)
- Adds the “reasonable person” standard;
- Key information (such as risks, benefits, side effects, and alternatives) must be provided upfront in a concise and focused manner;
- On the other hand, “concise” does not mean simply presenting a list of facts. Rather, the info should be well-organized and in sufficient detail to provide the context that a subject needs to fully understand and thus be able to make an informed decision about whether to participate in the research or not.
Informed Consent New Elements — Identifiable Private Information/Biospecimens
- (i) A statement that identifiers might be removed from the identifiable private information or identifiable biospecimens and that, after such removal, the information or biospecimens could be used for future research studies or distributed to another investigator for future research studies without additional informed consent from the subject or the legally authorized representative, if this might be a possibility; or
- (ii) A statement that the subject’s information or biospecimens collected as part of the research, even if identifiers are removed, will not be used or distributed for future research studies.
- (7) A statement that the subject’s biospecimens (even if identifiers are removed) may be used for commercial profit and whether the subject will or will not share in this commercial profit;
- (8) A statement regarding whether clinically relevant research results, including individual research results, will be disclosed to subjects, and if so, under what conditions; and
- (9) For research involving biospecimens, whether the research will (if known) or might include whole genome sequencing (i.e., sequencing of a human germline or somatic specimen with the intent to generate the genome or exome sequence of that specimen).
§ __.116(b)(9)(i)-(ii) and § __.116(c)(7)-(9)
§ __.116(b)(9)(i)-(ii) — Under the “Basic elements of informed consent” section:
Notice must be given to a subject that his or her identifiable private information or biospecimens may be stripped of identifiers and used in future research by the current investigator or distributed to another investigator without the subject’s informed consent, OR
Notice must be given to a subject that his or her information, even if de-identified, will not be used by any investigators for future research studies.
§ __.116(c)(7)-(9) — Under the “Additional elements of informed consent” section:
Notice must be given about:
- Possible commercial profit involving biospecimens and whether subject will share in this profit;
- Whether clinically relevant research results will be disclosed to subjects and under what conditions; and
- Whether the research involving biospecimens might include whole genome sequencing.
Elements of Broad Consent
Broad consent for the storage, maintenance, and secondary research use of identifiable private information or identifiable biospecimens (collected for either research studies other than the proposed research or nonresearch purposes) is permitted as an alternative to the informed consent requirements in paragraphs (b) and (c) of this section.
(1) The information required in paragraphs (b)(2) [foreseeable research-related risks or discomforts to subject], (b)(3) [research benefits to subject], (b)(5) [extent that confidentiality of records identifying the subject will be maintained], and (b)(8) [participation is voluntary and may be discontinued at any time without penalty or loss of benefits] and, when appropriate, (c)(7) [biospecimens may be used for commercial profit and whether subject will share in the profit] and (9) [biospecimen research may include whole genome sequencing] of this section;
(2) A general description of the types of research that may be conducted with the identifiable private information or identifiable biospecimens. This description must include sufficient information such that a reasonable person would expect that the broad consent would permit the types of research conducted;
(3) A description of the identifiable private information or identifiable biospecimens that might be used in research, whether sharing of identifiable private information or identifiable biospecimens might occur, and the types of institutions or researchers that might conduct research with the identifiable private information or identifiable biospecimens;
(4) A description of the period of time that the identifiable private information or identifiable biospecimens may be stored and maintained (which period of time could be indefinite), and a description of the period of time that the identifiable private information or identifiable biospecimens may be used for research purposes (which period of time could be indefinite);
(5) Unless the subject or legally authorized representative will be provided details about specific research studies, a statement that they will not be informed of the details of any specific research studies that might be conducted using the subject’s identifiable private information or identifiable biospecimens, including the purposes of the research, and that they might have chosen not to consent to some of those specific research studies;
(6) Unless it is known that clinically relevant research results, including individual research results, will be disclosed to the subject in all circumstances, a statement that such results may not be disclosed to the subject; and
(7) An explanation of whom to contact for answers to questions about the subject’s rights and about storage and use of the subject’s identifiable private information or identifiable biospecimens, and whom to contact in the event of a research-related harm.
§ __.116(d)(1)-(7)
Waivers
(e) Waiver or alteration of consent in research involving public benefit and service programs conducted by or subject to the approval of state or local officials.
(3) Requirements for waiver and alteration. In order for an IRB to waive or alter consent as described in this subsection, the IRB must find and document that:
(i) The research or demonstration project is to be conducted by or subject to the approval of state or local government officials and is designed to study, evaluate, or otherwise examine:
(A) Public benefit or service programs;
(B) Procedures for obtaining benefits or services under those programs;
(C) Possible changes in or alternatives to those programs or procedures; or
(D) Possible changes in methods or levels of payment for benefits or services under those programs; and
(ii) The research could not practicably be carried out without the waiver or alteration.
§ __.116(e)
(1) Waiver. If an individual was asked to provide broad consent for the storage, maintenance, and secondary research use of identifiable private information or identifiable biospecimens in accordance with the requirements at paragraph (d) of this section [see Elements of Broad Consent], and refused to consent, an IRB cannot waive consent for the storage, maintenance, or secondary research use of the identifiable private information or identifiable biospecimens.
(2) Alteration. An IRB may not omit or alter any of the requirements described in paragraph (a) of this section [“concise and focused presentation of key information .. in sufficient detail … to facilitate subject’s … understanding of reasons why one might or might not want to participate”].
If a broad consent procedure is used, an IRB may not omit or alter any of the elements required under paragraph (d) of this section [broad consent].
(3) Requirements for waiver and alteration. In order for an IRB to waive or alter consent as described in this subsection, the IRB must find and document that:
The research involves no more than minimal risk to the subjects;
(ii) The research could not practicably be carried out without the requested waiver or alteration;
(iii) If the research involves using identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format; [new criterion]
(iv) The waiver or alteration will not adversely affect the rights and welfare of the subjects; and
(v) Whenever appropriate, the subjects or legally authorized representatives will be provided with additional pertinent information after participation.
§ __.116(f)(1)-(3)
An IRB may approve a research proposal in which an investigator will obtain information or biospecimens for the purpose of screening, recruiting, or determining the eligibility of prospective subjects without the informed consent of the prospective subject or the subject’s legally authorized representative, if either of the following conditions are met:
(1) The investigator will obtain information through oral or written communication with the prospective subject or legally authorized representative, or
(2) The investigator will obtain identifiable private information or identifiable biospecimens by accessing records or stored identifiable biospecimens.
§ __.116(g)

Posting of Consent Forms for Clinical Trials
(1) For each clinical trial conducted or supported by a Federal department or agency, one IRB- approved informed consent form used to enroll subjects must be posted by the awardee or the Federal department or agency component conducting the trial on a publicly available Federal Web site that will be established as a repository for such informed consent forms.
(2) If the Federal department or agency supporting or conducting the clinical trial determines that certain information should not be made publicly available on a Federal Web site (e.g. confidential commercial information), such Federal department or agency may permit or require redactions to the information posted.
(3) The informed consent form must be posted on the Federal Web site after the clinical trial is closed to recruitment, and no later than 60 days after the last study visit by any subject, as required by the protocol.
§ __.116(h)
Documentation of Informed Consent (key points)
(a) Except as provided in paragraph (c) of this section, informed consent shall be documented by the use of a written informed consent form approved by the IRB and signed (including in an electronic format) by the subject or the subject’s legally authorized representative. A written copy shall be given to the person signing the informed consent form.
(2) A short form written informed consent form stating that the elements of informed consent required by § __.116 have been presented orally to the subject or the subject’s legally authorized representative, and that the key information required by § __.116(a)(5)(i) was presented first to the subject, before other information, if any, was provided.
(c)(1) An IRB may waive the requirement for the investigator to obtain a signed informed consent form for some or all subjects if it finds … the following:
(iii) If the subjects or legally authorized representatives are members of a distinct cultural group or community in which signing forms is not the norm, that the research presents no more than minimal risk of harm to subjects and provided there is an appropriate alternative mechanism for documenting that informed consent was obtained.
§ __.117
Limited IRB Review — New
Required for exemptions § __.104(d)(2)(iii), (d)(3)(i)(C), and (d)7, and (8) in the final, revised Common Rule to:
- protect the privacy of subjects and confidentiality of data under § __.111(a)(7) and
- ensure that conditions for broad consent involving identifiable private information and biospecimens are met.
§ __.109(a)
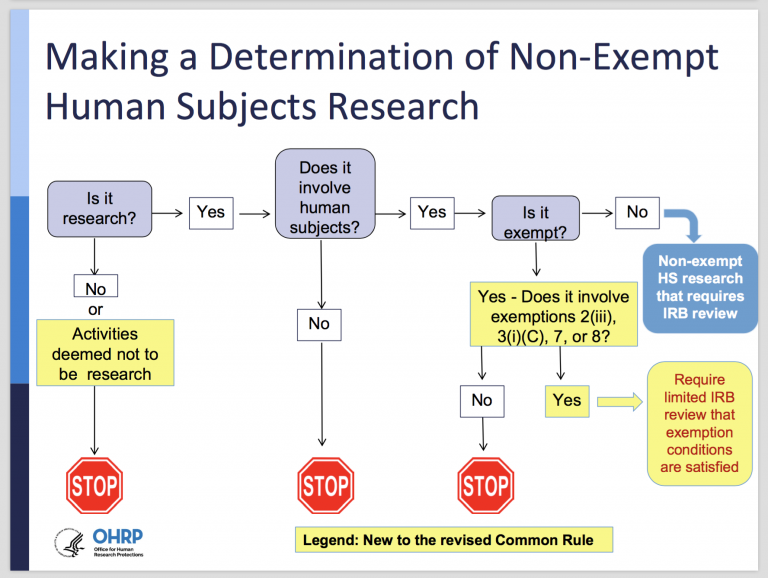
Expedited IRB Review — New
The Secretary of HHS has established, and published as a Notice in the Federal Register, a list of categories of research that may be reviewed by the IRB through an expedited review procedure. The Secretary will evaluate the list at least every 8 years and amend it, as appropriate, after consultation with other federal departments and agencies and after publication in the Federal Register for public comment.
(b)(1) An IRB may use the expedited review procedure to review the following:
(i) Some or all of the research appearing on the list described in paragraph (a) of this section, unless the reviewer determines that the study involves more than minimal risk;
(ii) Minor changes in previously approved research during the period for which approval is authorized; or
(iii) Research for which limited IRB review is a condition of exemption under § __.104(d)(2)(iii), (d)(3)(i)(C), and (d)(7) and (8).
(2) Under an expedited review procedure, the review may be carried out by the IRB chairperson or by one or more experienced reviewers designated by the chairperson from among members of the IRB. In reviewing the research, the reviewers may exercise all of the authorities of the IRB except that the reviewers may not disapprove the research. A research activity may be disapproved only after review in accordance with the nonexpedited procedure set forth in § __.108(b).
(c) Each IRB that uses an expedited review procedure shall adopt a method for keeping all members advised of research proposals that have been approved under the procedure.
(d) The department or agency head may restrict, suspend, terminate, or choose not to authorize an institution’s or IRB’s use of the expedited review procedure.
§ __.110 and § __.115(a)(8)
Continuing IRB Review — Changes
Unless an IRB determines otherwise, continuing review of research is not required in the following circumstances:
(i) Research eligible for expedited review in accordance with § __.110;
(ii) Research reviewed by the IRB in accordance with the limited IRB review described in § __.104(d)(2)(iii), (d)(3)(i)(C), or (d)(7) or (8);
(iii) Research that has progressed to the point that it involves only one or both of the following, which are part of the IRB-approved study:
(A) Data analysis, including analysis of identifiable private information or identifiable biospecimens, or
(B) Accessing follow-up clinical data from procedures that subjects would undergo as part of clinical care.
§ __.109(f) and § __.115(a)(3)
Single IRB Review Requirement
Any institution located in the United States that is engaged in cooperative research must rely upon approval by a single IRB for that portion of the research that is conducted in the United States. The reviewing IRB will be identified by the Federal department or agency supporting or conducting the research or proposed by the lead institution subject to the acceptance of the Federal department or agency supporting the research.
(2) The following research is not subject to this provision:
(i) Cooperative research for which more than single IRB review is required by law (including tribal law passed by the official governing body of an American Indian or Alaska Native tribe); or
(ii) Research for which any Federal department or agency supporting or conducting the research determines and documents that the use of a single IRB is not appropriate for the particular context.
§ __.114(b)
Got Questions?
UNC IRB and Office of Human Research Ethics
HHS.gov Office of Human Research Protections
Need a Quick Guide?
PRIM&R Final Common Rule Quick Reference Guide
Want More In-depth Info?
Common Rule-specific Definitions:
RESEARCH means a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.
4 activities NOT deemed research:
(1) Scholarly and journalistic activities (e.g., oral history, journalism, biography, literary criticism, legal research, and historical scholarship), including the collection and use of information, that focus directly on the specific individuals about whom the information is collected.
(2) Public health surveillance activities, including the collection and testing of information or biospecimens, conducted, supported, requested, ordered, required, or authorized by a public health authority. Such activities are limited to those necessary to allow a public health authority to identify, monitor, assess, or investigate potential public health signals, onsets of disease outbreaks, or conditions of public health importance (including trends, signals, risk factors, patterns in diseases, or increases in injuries from using consumer products). Such activities include those associated with providing timely situational awareness and priority setting during the course of an event or crisis that threatens public health (including natural or man-made disasters).
(3) Collection and analysis of information, biospecimens, or records by or for a criminal justice agency for activities authorized by law or court order solely for criminal justice or criminal investigative purposes.
(4) Authorized operational activities (as determined by each agency) in support of intelligence, homeland security, defense, or other national security missions.
HUMAN SUBJECT means a living individual about whom an investigator (whether professional or student) conducting research:
- Obtains information or biospecimens through intervention or interaction with the individual, AND uses, studies, or analyzes the information or biospecimens; or
- Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
IDENTIFIABLE PRIVATE INFORMATION is private information for which the identity of the subject is or may readily be ascertained by the investigator or associated with the information.
An IDENTIFIABLE BIOSPECIMEN is a biospecimen for which the identity of the subject is or may readily be ascertained by the investigator or associated with the biospecimen.
Federal departments or agencies implementing this policy shall:
Upon consultation with appropriate experts (including experts in data matching and re-identification), reexamine the meaning of ‘‘identifiable private information’’ and ‘‘identifiable biospecimen,’’ as defined in this section. This reexamination shall take place within 1 year and regularly thereafter (at least every 4 years). This process will be conducted by collaboration among the Federal departments and agencies implementing this policy. If appropriate and permitted by law, such Federal departments and agencies may alter the interpretation of these terms, including through the use of guidance.
Upon consultation with appropriate experts, assess whether there are analytic technologies or techniques that should be considered by investigators to generate ‘‘identifiable private information,’’ or an ‘‘identifiable biospecimen,’’ as defined in this section. This assessment shall take place within 1 year and regularly thereafter (at least every 4 years). This process will be conducted by collaboration among the Federal departments and agencies implementing this policy.
Any such technologies or techniques will be included on a list of technologies or techniques that produce identifiable private information or identifiable biospecimens. This list will be published in the Federal Register after notice and an opportunity for public comment. The Secretary, HHS, shall maintain the list on a publicly accessible Web site.
INTERVENTION includes both physical procedures by which information or biospecimens are gathered (e.g., venipuncture) and manipulations of the subject or the subject’s environment that are performed for research purposes.
INTERACTION includes communication or interpersonal contact between investigator and subject.
MINIMAL RISK means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
Research involving BENIGN BEHAVIORAL INTERVENTIONS in conjunction with the collection of information from an adult subject through verbal or written responses (including data entry) or audiovisual recording if the subject prospectively agrees to the intervention and information collection and at least one of the following criteria is met:
(A) The information obtained is recorded by the investigator in such a manner that the identity of the human subjects cannot readily be ascertained, directly or through identifiers linked to the subjects;
(B) Any disclosure of the human subjects’ responses outside the research would not reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, educational advancement, or reputation; or
(C) The information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects, and an IRB conducts a limited IRB review to make the determination required by § __.111(a)(7).
- For the purpose of this provision, BENIGN BEHAVIORAL INTERVENTIONS are brief in duration, harmless, painless, not physically invasive, not likely to have a significant adverse lasting impact on the subjects, and the investigator has no reason to think the subjects will find the interventions offensive or embarrassing. Provided all such criteria are met, examples of such benign behavioral interventions would include having the subjects play an online game, having them solve puzzles under various noise conditions, or having them decide how to allocate a nominal amount of received cash between themselves and someone else.
- If the research involves deceiving the subjects regarding the nature or purposes of the research, this exemption is not applicable unless the subject authorizes the deception through a prospective agreement to participate in research in circumstances in which the subject is informed that he or she will be unaware of or misled regarding the nature or purposes of the research.
SECONDARY RESEARCH for which consent is not required: Secondary research uses of identifiable private information or identifiable biospecimens, if at least one of the following criteria is met:
- The identifiable private information or identifiable biospecimens are publicly available;
- Information, which may include information about biospecimens, is recorded by the investigator in such a manner that the identity of the human subjects cannot readily be ascertained directly or through identifiers linked to the subjects, the investigator does not contact the subjects, and the investigator will not re-identify subjects;
- The research involves only information collection and analysis involving the investigator’s use of identifiable health information when that use is regulated under 45 CFR parts 160 and 164, subparts A and E, for the purposes of ‘‘health care operations’’ or ‘‘research’’ as those terms are defined at 45 CFR 164.501 or for ‘‘public health activities and purposes’’ as described under 45 CFR 164.512(b); or
- The research is conducted by, or on behalf of, a Federal department or agency using government-generated or government-collected information obtained for nonresearch activities, if the research generates identifiable private information that is or will be maintained on information technology that is subject to and in compliance with [various federal privacy laws].
PUBLIC HEALTH AUTHORITY refers to an agency or authority of the United States, a state, a territory, a political subdivision of a state or territory, an Indian tribe, or a foreign government, or a person or entity acting under a grant of authority from or contract with such public agency, including the employees or agents of such public agency or its contractors or persons or entities to whom it has granted authority, that is responsible for public health matters as part of its official mandate.
CLINICAL TRIAL refers to a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) in order to evaluate the effects of the interventions on biomedical or behavioral health-related outcomes.
LEGALLY AUTHORIZED REPRESENTATIVE refers to an individual, judicial, or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s participation in the procedure(s) involved in the research.
If there is no applicable law addressing this issue, legally authorized representative means an individual recognized by institutional policy as acceptable for providing consent in the nonresearch context on behalf of the prospective subject to the subject’s participation in the procedure(s) involved in the research.